




开放科学(资源服务)标识码(OSID): 
天竺桂(Cinnamomum japonicum)为樟科(Lauraceae)樟属(Cinnamomum)常绿阔叶乔木,我国三级保护植物, 具有喜光、耐旱、速生等生长特性,多生于海拔300~1 000 m常绿阔叶林中,星散分布于中国、朝鲜和日本。众所周知,香樟(C. camphora)是一种园林绿化珍贵树种,然而,由于耐寒性不强,限制了其向长江以北及高海拔地区的推广,而同属乔木天竺桂,不仅具有香樟相近的优美树姿,同时具有更强的耐寒性,表现出较高的推广潜力,在园林绿化中的作用越来越受到重视。此外,天竺桂还具有重要的药用、生化和用材价值。由于中国天竺桂野生资源中蕴含着极具潜力的独特基因, 是天竺桂育种改良宝贵的基因资源, 也是我国重要的植物遗传资源。因此, 对天竺桂野生资源的研究与保护已经开始引起越来越多的重视。
近年来对天竺桂野生资源的考察发现,由于人类活动频繁和21世纪初对森林的大量砍伐, 许多天竺桂天然群体已经消失,现存的天竺桂天然群体也因生态环境的严重破坏而处于濒危状态[1]。天竺桂被中国植物物种信息数据库收录为国家三级保护树种。如何加强天竺桂资源的保护和利用亟待解决。因此, 开展天竺桂种质资源研究, 评估天然群体遗传多样性和群体遗传结构,将为制定科学有效的天竺桂资源保护策略提供重要参考,同时也是天竺桂遗传育种研究中不可缺少的基础性工作。然而, 迄今天竺桂分子生物学和遗传学研究基础十分薄弱,对其野生资源遗传多样性及群体遗传结构研究也鲜见报道。
利用分子标记技术进行群体遗传变异分析是群体遗传学研究的必要手段。常用于林木群体遗传学研究的分子标记包括简单序列重复[2⇓-4]、扩增片段长度多态性标记[5]、随机扩增多态性DNA[4]等。传统开发标记的方法,获得的标记数量少,不具有代表性,而且需要花费大量的人力和时间。简化基因组测序是基于高通量测序平台的大规模分子标记开发的方法。相对于传统的标记开发方法,具有高通量、自动化、准确度高等优点,此外还可以同时获得分子标记和基因分型数据,已成为最受欢迎的群体遗传学研究手段[6]。特异位点扩增片段测序(specific-locus amplified fragment sequencing, SLAF-seq)技术是简化基因组测序的一种,近年来,在无参物种的群体遗传学研究中获得广泛应用。陈立杰等[7]利用SLAF-seq技术从48份贵州古茶树资源中获得了283 376个高度一致性的群体单核苷酸多态性(single nucleotide polymorphism, SNP)标记,基于这些SNP的群体结构分析可清晰地把古茶树资源分为乔木型和灌木型2个类群。段义忠等[8]利用SLAF-seq技术开发获得102 025 个高度一致性的沙冬青(Ammopiptanthus mongolicus)群体 SNP,并进行10 个地点的沙冬青的遗传分析。可见,SLAF-seq已成为无参物种群体遗传学研究中的一项可靠工具。本研究拟利用SLAF-seq技术进行天竺桂的大规模SNP标记开发及遗传分析,为我国天竺桂资源的有效利用与科学保护奠定理论基础。
1 材料与方法
1.1 植物材料
以2018年自5省(直辖市)的7个天然分布区采集的30份天竺桂叶片样品为研究材料。其中,6份采自浙江省建德市莲花镇(编号为ZJ-LHZ)(119°18'58″E,29°34'20″N),3份采自浙江省建德市天目山(ZJ-TMS) (119°17'32″E,29°31'18″N),3份来自安徽省宣城市泾县桃岭(AH-TL)(118°35'38″E,30°29'35″N),5份来自安徽省霍山县(AN-HS)(116°13'44″E,31°20'2″N),5份来自河南省洛阳市栾川县伏牛山(HN-FNS)(111°41'29″E,33°43'44″N),5份采自重庆市万州区(CQ-WZ)(108°25'9″E、30°49'30″N),5份采自云南省昆明市滇池捞鱼河湿地公园(YN-DC)(102°46'41″E,24°49'43″N)。
1.2 植物DNA提取
采用百泰克生物技术有限公司(北京)新型快速植物基因组提取试剂盒提取叶片基因组DNA。利用伯托生物技术有限公司(德国)Berthold colibri超微量分光光度计检测DNA的浓度和纯度,将质量浓度稀释至25 ng/μL,置于-20 ℃冰箱中备用。
1.3 建库与测序
选取天竺桂近缘种沉水樟(C. micranthum)基因组作为参考基因组进行酶切预测。沉水樟基因组组装出的基因组大小为730 Mb,GC含量为38.34%(ftp://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/003/546/025/GCA_003546025.1_ASBRC_Ckan_1.0/)。利用百迈克生物技术有限公司自主研发的酶切预测软件对参考基因组进行酶切预测[9]。最终确定使用Rsa I+HinC Ⅱ对检测合格的各样品基因组DNA分别进行酶切。将酶切片段长度在264~314 bp的序列定义为SLAF标签,预测将获得131 530个SLAF标签。SLAF标签进行3'端加A处理、连接Dual-index[10]测序接头、PCR扩增、纯化、混样、切胶选取目的片段,文库质检合格后用Illumina平台进行测序。建库、测序工作委托北京百迈克生物技术有限公司进行。
为评估酶切实验的准确性,选用水稻‘日本晴’(Oryza sativa subsp. japonica cv. Nipponbare)作为对照,其基因组大小为374.30 Mb (
1.4 SLAF标签开发及SNP信息统计
经过SLAF-seq建库产生的特定酶切片段,一个酶切片段就是一个SLAF标签。本项目测序产生的reads来源于同一限制性内切酶对不同样品作用产生的长度相同的酶切片段,根据序列相似性将各样品的reads进行聚类,聚类到一起的reads来源于一个SLAF片段(SLAF标签)。同一SLAF标签在不同样品间的序列相似度远高于不同SLAF标签间的相似度;一个SLAF标签在不同样品间序列有差异(即有多态性),即可定义为多态性SLAF标签。
1.5 遗传多样性分析
1.6 遗传进化分析
2 结果与分析
2.1 建库测序结果
对照水稻‘日本晴’测序获得总reads数为749 149,GC含量为41.39%,Q30为93.43%。通过Soap[20]软件将对照水稻‘日本晴’基因组Rsa Ⅰ+HinC Ⅱ的酶切后的测序reads与其参考基因组进行比对,双端比对效率为94.78%。统计水稻‘日本晴’测序reads插入片段中残留酶切位点的比例为89.93%,表明酶切效率较高。水稻‘日本晴’数据的双端比对效率和酶切效率评估结果均说明本次SLAF建库正常。
30个天竺桂样品测序后共获得100.78 Mb reads数据,为保证分析质量,采用读长126 bp×2作为后续的数据评估和分析数据。各样品测序质量值(Q)在92.81%~94.04%之间,表明测序碱基测序错误率低。各样品的碱基分布均显示出reads的前2个碱基呈现与酶切位点一致的碱基分离,后续碱基分布会呈现不同程度的小幅波动,复合DNA酶切片段的碱基分布特征。对各样品的测序数据reads数量、Q30和GC含量进行统计,各样品reads数在1 367 794~5 927 25之间。GC含量为39.14%~41.73%,平均GC含量为40.05%,平均Q30为93.51%。
2.2 SLAF标记及SNP信息统计结果
根据序列相似性将各样品的reads进行聚类,共获得1 296 000个SLAF标签,各样品所含的SLAF标签数为135 077~215 578个,各样品匹配到SLAF标签的总reads数为1 080 909~5 157 597,各样品的测序深度为7.68~23.92倍,平均测序深度为15.59倍。其中377 250个多态性SLAF标签在不同样品间序列有差异。
以每个SLAF标签中测序深度最高的序列类型作为参考序列开发SNP标记,共得到3 409 402个群体SNP,各样品中检测到的SNP个数为917 896~1 386 854,各样品中检测到的SNP杂合率为5.61%~9.07%,各样品中SNP的完整度为26.92%~40.67%(表1)。
表1 30个样品的SNP信息统计
Table 1
| 样品编号 sample ID | SNP个数 SNP number | SNP 杂合率/% hetloci ratio | SNP 完整度/% integrity ratio |
|---|---|---|---|
| ZJ-LHZ-1 | 1 103 214 | 6.74 | 32.35 |
| ZJ-LHZ-2 | 1 042 601 | 6.34 | 30.58 |
| ZJ-LHZ-3 | 1 114 298 | 6.95 | 32.68 |
| ZJ-LHZ-4 | 1 138 102 | 7.16 | 33.38 |
| ZJ-LHZ-5 | 1 116 854 | 7.08 | 32.75 |
| ZJ-LHZ-6 | 944 920 | 5.68 | 27.71 |
| ZJ-TMS-3 | 1 320 574 | 7.48 | 38.73 |
| ZJ-TMS-4 | 1 373 270 | 7.66 | 40.27 |
| ZJ-TMS-5 | 1 211 864 | 8.43 | 35.54 |
| AH-JX-1 | 1 368 810 | 8.17 | 40.14 |
| AH-JX-2 | 917 896 | 6.25 | 26.92 |
| AH-JX-3 | 1 004 978 | 6.44 | 29.47 |
| AH-HS-1 | 1 352 623 | 8.98 | 39.67 |
| AH-HS-2 | 1 259 826 | 8.52 | 36.95 |
| AH-HS-3 | 1 386 854 | 8.30 | 40.67 |
| AH-HS-4 | 1 367 363 | 9.07 | 40.10 |
| AH-HS-5 | 1 345 653 | 8.91 | 39.46 |
| HN-FNS-1 | 1 249 145 | 8.12 | 36.63 |
| HN-FNS-2 | 1 337 990 | 8.44 | 39.24 |
| HN-FNS-3 | 1 229 537 | 8.72 | 36.06 |
| CQ-WZ-1 | 1 062 854 | 5.80 | 31.17 |
| CQ-WZ-2 | 1 013 706 | 6.20 | 29.73 |
| CQ-WZ-3 | 1 089 181 | 5.78 | 31.94 |
| CQ-WZ-4 | 1 022 760 | 5.61 | 29.99 |
| CQ-WZ-5 | 987 065 | 5.81 | 28.95 |
| YN-DC-1 | 986 326 | 5.64 | 28.92 |
| YN-DC-2 | 1 119 619 | 5.95 | 32.83 |
| YN-DC-3 | 1 012 986 | 5.83 | 29.71 |
| YN-DC-4 | 1 095 620 | 5.67 | 32.13 |
| YN-DC-5 | 1 012 025 | 5.89 | 29.68 |
根据完整度>80%,次要基因型频率>0.05过滤,共得到268 821个高一致性的群体SNP用于后续的遗传进化相关分析。
2.3 天竺桂7个群体的遗传多样性分析
由7个群体遗传群体多样性情况(表2)可知,平均有效等位基因数为1.222 3(1.180 5~1.277 5)个,Nei(h)指数平均为0.148 9(0.117 8~0.180 4) Shannon指数(I)平均为0.190 9(0.156 3~0.240 2),根据Nei(h)和Shannon指数7个群体的遗传多样性由高到低依次为:AH-HS、ZJ-LHZ、ZJ-TMS、AH-JX、HN-FNS、YN-DC和CQ-WZ。7个群体的遗传多样性相近,Nei(h)指数均较小,表明7个群体均具有较低的遗传多样性,这可能与群体采集的个体较少有关。
表2 天竺桂7个群体遗传多样性比较
Table 2
| 群体 population | Ne | Ho | He | h | I | PIC | Fis |
|---|---|---|---|---|---|---|---|
| AH-HS | 1.277 5 | 0.171 7 | 0.161 4 | 0.180 4 | 0.240 2 | 0.128 8 | 0.408 8 |
| AH-JX | 1.214 3 | 0.143 0 | 0.122 1 | 0.149 3 | 0.178 7 | 0.096 4 | 0.512 5 |
| CQ-WZ | 1.180 5 | 0.104 7 | 0.106 0 | 0.119 1 | 0.159 0 | 0.085 0 | 0.637 0 |
| HN-FNS | 1.208 1 | 0.127 8 | 0.118 1 | 0.142 6 | 0.172 6 | 0.093 1 | 0.563 2 |
| YN-DC | 1.181 4 | 0.099 9 | 0.105 1 | 0.117 8 | 0.156 3 | 0.083 8 | 0.655 6 |
| ZJ-LHZ | 1.263 2 | 0.133 3 | 0.155 8 | 0.172 0 | 0.234 3 | 0.125 3 | 0.533 1 |
| ZJ-TMS | 1.231 1 | 0.141 7 | 0.132 6 | 0.161 1 | 0.194 9 | 0.105 1 | 0.516 8 |
| 平均mean | 1. 222 3 | 0.131 7 | 0. 128 7 | 0.148 9 | 0.190 9 | 0.102 5 | 0.546 7 |
7个群体平均观察杂合度(Ho)为0.131 7(0.099 9~0.143 0),期望杂合度(He)为0.128 7(0.105 1~0.161 4),观察杂合度与期望杂合度基本相近,表明该群体中的杂合体与纯合体基本相当,较接近随机交配。
采用Nei的F统计量(Fst)对群体的遗传变异进行分析,确定位点上的变异量在群体间及群体内的分布情况(表3)。7个群体两两间的基因分化系数(Fst)存在显著差异,为0.024 2~0.686 3,其中CQ-WZ和YN-DC群体的基因分化系数最小(0.024 2),即2.42%的遗传变异存在于群体间,97.58%存在于群体内,表明两群体间的遗传分化较小。AH-JX和YN-DC群体的基因分化系数最大(0.686 3),即遗传变异68.63%存在于群体间,31.37%存在于群体内,表明两群体间存在较大的遗传变异。
表3 群体间的F统计量计算
Table 3
| 比较组 comparable groups | Fst |
|---|---|
| AH-HS vs AH-JX | 0.265 6 |
| AH-HS vs CQ-WZ | 0.639 2 |
| AH-HS vs HN-FNS | 0.360 7 |
| AH-HS vs YN-DC | 0.643 1 |
| AH-HS vs ZJ-LHZ | 0.220 6 |
| AH-HS vs ZJ-TMS | 0.242 7 |
| AH-JX vs CQ-WZ | 0.682 7 |
| AH-JX vs HN-FNS | 0.450 1 |
| AH-JX vs YN-DC | 0.686 3 |
| AH-JX vs ZJ-LHZ | 0.189 0 |
| AH-JX vs ZJ-TMS | 0.219 9 |
| CQ-WZ vs HN-FNS | 0.658 8 |
| CQ-WZ vs YN-DC | 0.024 2 |
| CQ-WZ vs ZJ-LHZ | 0.654 6 |
| CQ-WZ vs ZJ-TMS | 0.667 9 |
| HN-FNS vs YN-DC | 0.662 2 |
| HN-FNS vs ZJ-LHZ | 0.406 7 |
| HN-FNS vs ZJ-TMS | 0.428 3 |
| YN-DC vs ZJ-LHZ | 0.658 4 |
| YN-DC vs ZJ-TMS | 0.671 7 |
| ZJ-LHZ vs ZJ-TMS | 0.148 6 |
2.4 天竺桂7个群体的遗传进化分析
基于过滤后得到的268 821个高一致性的群体SNP,对来自5省(市)7个天然群体的30份天竺桂资源进行系统发育分析、群体遗传结构分析和主成分分析(PCA),从多个方面揭示天竺桂的遗传分化关系。
2.4.1 系统发育分析
来自7个群体的30份天竺桂资源的进化树如图1所示。7个天竺桂群体起源的地理分布由东至西依次为浙江莲花镇(119°18'58″E)—浙江天目山(119°17'32″E)—安徽桃岭(118°35'38″E)—安徽霍山 (116°13'44″E)—河南伏牛山(111°41'29″E)—重庆万州(108°25'9″E)—云南滇池(102°46'41″E)。从进化树可以看出,来自同一群体的个体具有较近的亲缘关系,群体内变异小于群体间变异。位于进化树最下端的天竺桂为来自浙江莲花镇(ZJ-LHZ)的6份天竺桂资源。顺着进化树向上为来自浙江天目山(ZJ-TMS)的3份、来自安徽桃岭(AH-TL)的3份、来自安徽霍山(AH-HS)的5份、河南伏牛山(HN-FNS)的3份,顺着进化树继续向上,为重庆万州(CQ-WZ)的5份,最上端的天竺桂资源分别为来自云南滇池(YN-DC)的5份。即7个群体的天竺桂资源的进化关系依次为ZJ-LHZ、ZJ-TMS、AH-TL、AH-HS、HN-FNS、CQ-WZ和YN-DC,与它们在地理经度上的远近关系一致。天竺桂亲缘关系与地理分布格局显示出一致性,呈现由东向西的进化趋势,可见,天竺桂是由中国东部向西逐渐进化的。
图1
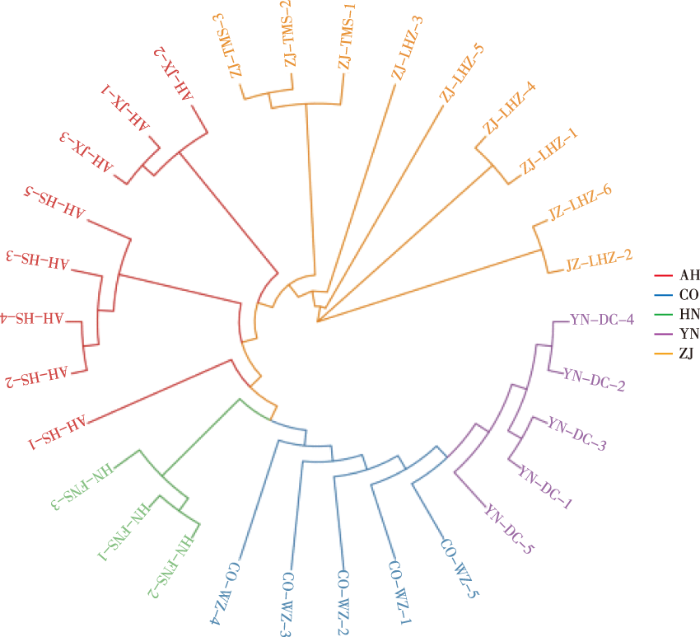
图1
30份供试种质材料的遗传进化树
橙色(浙江种源) orange(Zhejiang provenance);紫色(云南种源) purple (Yunnan provenance);蓝色(重庆种源) blue(Chongqing provenance);绿色(河南种源) green (Henan provenance);红色(安徽种源) red (Anhui provenance)。
Fig.1
Evolutionary tree of the 30 test materials
2.4.2 遗传结构与主成分分析
图2
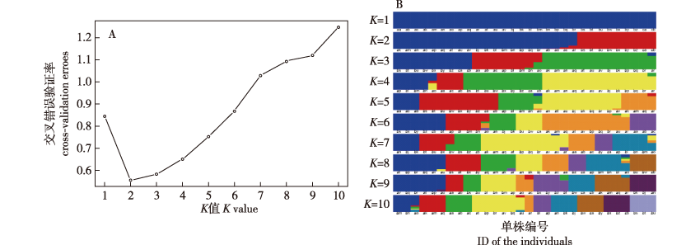
图2
不同K值所对应的交叉验证错误率及个体聚类图
Fig.2
The admixture validation error rate and individual cluster values corre-sponding to the different K values
PCA结果(图3)显示,来自浙江省的两个群体浙江莲花镇镇(ZJ-LHZ)和浙江天目山(ZJ-TMS)的9份天竺桂资源紧密地聚集在一起,来自安徽桃岭(AH-TL)的3份资源聚成一簇,并与来自浙江的9份资源在PCA图上分布紧密重叠。来自安徽霍山(AH-HS)的5份资源聚成另外一簇,与前一簇距离比较疏远。来自河南的3份天竺桂资源位于PCA图的左上侧,并不紧密聚集。来自重庆的5份天竺桂资源和来自云南5份的天竺桂资源在PCA上的分布紧密重叠,位于PCA图最右侧,而与其他群体的天竺桂资源距离较远。
图3
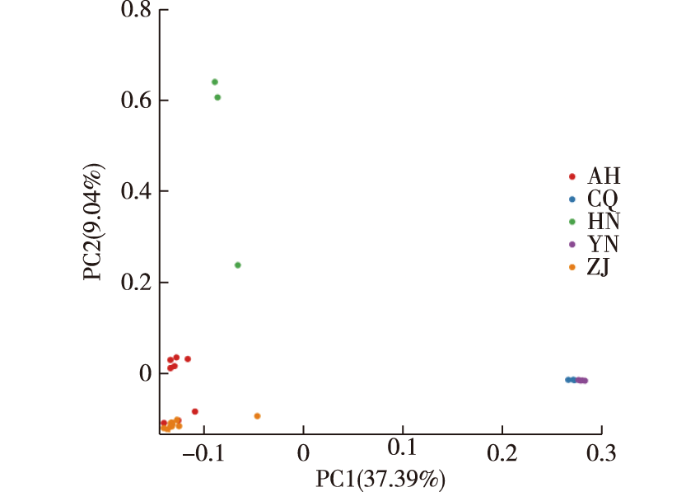
PCA分析结果显示,来自浙江省内的天竺桂资源遗传变异较小。然而,来自安徽桃岭(AH-TL)的3份资源聚成一簇,并与来自浙江的9份资源基本聚在一起,来自安徽霍山(AH-HS)的5份资源单独聚成另一簇,并于安徽桃岭(AH-TL)的资源在PCA图上存在一定的距离,说明,安徽省内的天竺桂资源存在较大的遗传变异。来自河南伏牛山的天竺桂群体内存在一定的遗传变异,但仍小于与其他群体间的遗传变异。来自重庆和云南的10份天竺桂资源紧密聚集,而与其他群体的天竺桂资源距离疏远,说明这两个群体间遗传变异较小,且与其他群体的天竺桂资源亲缘关系较远,这与群体结构分析的结果一致。
3 讨论
我国西部海拔高,东部海拔低,地势由西向东呈3级阶梯状逐级下降,其中第1阶梯平均海拔在4 000 m以上,第2阶梯平均海拔在1 000~2 000 m,第3阶梯平均海拔在500 m以下。其中,云南和重庆属于第2阶梯,安徽、浙江和河南属于第3阶梯。第2阶梯和第3阶梯之间以大兴安岭—太行山脉—巫山—雪峰山为分界线,因此,高大的山脉阻隔可能阻隔了第2、3阶梯之间天竺桂的基因交流,使得山脉两侧的天竺桂群体的基因频率发生改变,加上受不同海拔环境的选择,使得山脉两侧的天竺桂种群向不同方向发展,并形成了2个亚群。
安徽省内的天竺桂资源存在较大的遗传变异。这可能是因为长江位于安徽桃岭和霍山之间,对长江以北的霍山群体(AH-HS)与长江以南的泾县群体(AH-TL)间的基因交流产生阻隔,而泾县群体与同样位长江以南的浙江群体间存在更加频繁的基因交流。
系统进化、主成分分析和群体结构分析均显示,来自5省(市)7个天然群体的30份天竺桂资源可分为2个大的亚群,分别位于中国第2和第3阶梯上,2个亚群间被大兴安岭—太行山脉—巫山—雪峰山山脉阻隔。其中来自重庆和云南的天竺桂资源间遗产变异较小,且与其他群体间的亲缘关系较为疏远。而位于第3阶梯上的另一亚群,进一步可分为3个小亚群,其中来自浙江省和安徽桃岭的为第1个小亚群,来自安徽霍山的为第2个小亚群,而来自河南伏牛山的为第3个小亚群,其中,第1个和第2个小亚群间被长江阻隔。
本研究表明,天竺桂是由中国东部向中国西部逐渐进化的。山脉和湖泊阻隔带来的地理隔离是造成天竺桂资源遗传的重要原因,促进不同亚群间的基因交流 有助于保护天竺桂的遗传多样性。本研究首次揭示天竺桂遗传结构及地理变化规律,为我国天竺桂资源的有效利用与科学保护提供理论依据。
参考文献
栾川县野生天竺桂资源保护初探
[J].
Preliminary study on the protection of wild Cinnamomum japonicum resources in Luanchuan County
[J].
落羽杉属种类及其杂交子代‘中山杉’系列品种的SSR指纹图谱构建及遗传关系分析
[J].
Construction of SSR fingerprint and analysis on genetic relationship of Taxodium species and their hybrid progenies T.‘Zhongshanshan’series cultivars
[J].
Genetic diversity analysis of Sinojackia microcarpa,a rare tree species endemic in China,based on simple sequence repeat markers
[J].
Genetic diversity and differentiation of pedunculate (Quercus robur) and sessile (Q. petraea) oaks
[J].
Range-wide genetic diversity in natural populations of Larix principis-rupprechtii Mayr
[J].
基于RAD-seq技术的鹅掌楸基因组SNP标记开发
[J].
Development of genomic SNP markers based on RAD-seq and genome data in Liriodendron
[J].
贵阳花溪古茶树遗传进化的SNP分析
[J].
SNP analysis of the genetic evolution of ancient Camellia sinensis trees from Huaxi,Guiyang
[J].
基于SLAF-seq简化基因组技术的沙冬青SNP位点开发及遗传分析
[J].
SNP sites developed by specific length amplification fragment sequencing(SLAF-seq) and genetic analysis in Ammopitanthus mongolicus
[J].
Special features of RAD Sequencing data: implications for genotyping
[J].
Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform
[J].
Fast and accurate long-read alignment with Burrows-Wheeler transform
[J].
The genome analysis toolkit:a MapReduce framework for analyzing next-generation DNA sequencing data
[J].
The sequence alignment/map format and SAMtools
[J].
The variant call format and VCFtools
[J].
PopGenome:an efficient Swiss army knife for population genomic analyses in R
[J].
MEGA X:Molecular evolutionary genetics analysis across computing platforms
[J].
Fast model-based estimation of ancestry in unrelated individuals
[J].
Inference of population structure using multilocus genotype data
[J].
Principal components analysis corrects for stratification in genome-wide association studies
[J].
SOAP2: an improved ultrafast tool for short read alignment
[J].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}