 PDF(6238 KB)
PDF(6238 KB)

Identification and expression analysis of the bHLH gene family in Nitraria sibirica
ZHANG Jianjun, WU Jingxiang, FANG Hao, ZHU Liming, CHENG Tielong
Journal of Nanjing Forestry University (Natural Sciences Edition) ›› 2025, Vol. 49 ›› Issue (6) : 102-114.
PDF(6238 KB)
 PDF(6238 KB)
PDF(6238 KB)
Identification and expression analysis of the bHLH gene family in Nitraria sibirica
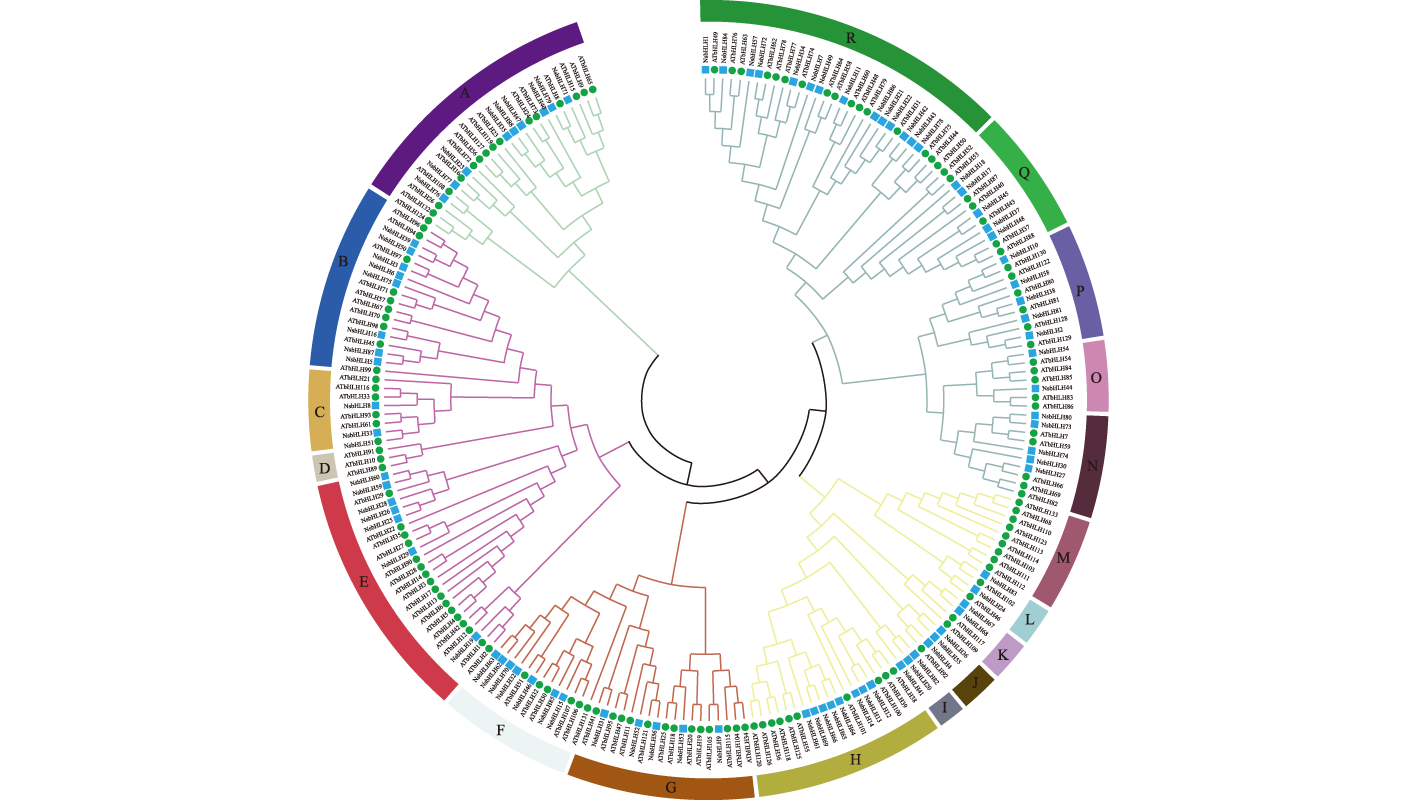
【Objective】The basic helix-loop-helix (bHLH) transcription factors play critical roles in plant growth, metabolism, and responses to biotic/abiotic stresses. Genome-wide identification and bioinformatics analysis of the bHLH gene family in Nitraria sibirica will provide foundational insights for subsequent bHLH gene cloning and further biological studies.【Method】Bioinformatics and molecular biology approaches were employed to systematically analyze the phylogenetic relationships of the N. sibirica bHLH gene family at the genome-wide level. Transcriptome datasets and quantitative reverse transcription-PCR (qRT-PCR) were employed to analyze the response of the bHLH genes under to stress.【Result】A total of 88 bHLH transcription factors were identified from the N. sibirica genome, unevenly distributed across 12 chromosomes with two pairs of tandemly duplicated gene. Phylogenetic analysis classified these members into 16 distinct subclades. Cis-acting element analysis revealed that the 88 bHLH genes harbored multiple environment- and stress-responsive regulatory elements. Expression profiling demonstrated differential responses of N. sibirica bHLH genes to salt stress. qRT-PCR results further demonstrated that six bHLH genes (bHLH1, bHLH2, bHLH24, bHLH30, bHLH70 and bHLH73) exhibited pronounced tissue-specific expression patterns. Under salt stress, bHLH1, bHLH24, bHLH70 and bHLH73 were significantly upregulated in leaves, while bHLH2 and bHLH30 were down regulated in leaves. In roots, all six genes displayed marked upregulation. In stems, five genes (bHLH1, bHLH24, bHLH30, bHLH70 and bHLH73) were upregulated, whereas bHLH2 showed down regulation.【Conclusion】This study identified 88 conserved bHLH transcription factors in N. sibirica, characterized by conserved gene structures and protein domains, along with two tandem duplication events on chromosomes. The differential expression of bHLH genes in roots, stems and leaves under salt stress suggests their potential involvement in salt tolerance mechanisms. These findings establish a critical foundation for elucidating the functional roles of the bHLH transcription factor family in N. sibirica.
Nitraria sibirica / bHLH transcription factors / salt stress / gene expression
| [1] |
朱涛, 李芳菲, 杨海涵, 等. 山药bHLH基因家族鉴定及表达分析[J]. 信阳师范学院学报(自然科学版), 2022, 35(3):393-399.
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
任镘蓉, 全英杰, 杨雯婷, 等. 地被菊bHLH转录因子基因家族鉴定及其低温胁迫响应[J]. 农业生物技术学报, 2022, 30(1):50-62.
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
何洁, 顾秀容, 魏春华, 等. 西瓜bHLH转录因子家族基因的鉴定及其在非生物胁迫下的表达分析[J]. 园艺学报, 2016, 43(2):281-294.
|
| [18] |
耿晶晶. 甜橙bHLH家族转录因子发掘及CsbHLH18抗寒功能鉴定与作用机制解析[D]. 武汉: 华中农业大学, 2018.
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
李焕勇, 杨秀艳, 唐晓倩, 等. NaCl处理对小果白刺叶片主要渗透调节物质和激素水平的影响[J]. 东北林业大学学报, 2019, 47(5):30-35.
|
| [23] |
胡娟, 张洁, 盛洲, 等. 不同预处理对柴达木盆地高寒湿地小果白刺种子萌发的影响[J]. 种子, 2022, 41(3):87-92,2.
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
尚先文, 范付华, 周紫晶, 等. 马尾松苗期转录组bHLH基因家族成员鉴定及表达分析[J]. 农业生物技术学报, 2020, 28(11):1947-1959.
|
| [33] |
赵安琪, 张世杰, 张志国, 等. 海水胁迫下萱草bHLH转录因子家族的鉴定及表达分析[J]. 分子植物育种, 2025, 23(16):5332-5343.
|
| [34] |
陈微, 潘美红, 惠林冲, 等. 基于转录组的洋葱bHLH转录因子家族鉴定与生物信息学分析[J]. 江苏农业科学, 2023, 51(5):11-18.
|
| [35] |
廖桂花, 段钰, 王丛丛, 等. 赪桐bHLH转录因子家族鉴定及生物信息学分析[J/OL]. 分子植物育种, 2023: 1-17. (2025-03-15).
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
冯建英, 李立芹, 鲁黎明. 马铃薯bHLH转录因子家族全基因组鉴定与表达分析[J]. 生物技术通报, 2022, 38(2):21-33.
|
| [40] |
张尚宏, 屈良鹄. 基因组的进化与内含子中的基因的进化[J]. 中山大学学报(自然科学版), 1999, 38(1):49-53.
|
| [41] |
|
| [42] |
|
| [43] |
高莉娟, 张正社, 文裕, 等. 象草全基因组bHLH转录因子家族鉴定及表达分析[J]. 草业学报, 2022, 31(3):47-59.
|
| [44] |
|
| [45] |
|
| [46] |
|
/
| 〈 |
|
〉 |





