 PDF(2336 KB)
PDF(2336 KB)

Genetic diversity and genetic structure analysis of principal Pseudotaxus chienii natural populations in Zhejiang Province
CAO Sen, GAO Kai, LIU Lingjuan, FANG Wanli, HE Xuekai, ZHOU Zhichun
Journal of Nanjing Forestry University (Natural Sciences Edition) ›› 2025, Vol. 49 ›› Issue (6) : 115-124.
PDF(2336 KB)
 PDF(2336 KB)
PDF(2336 KB)
Genetic diversity and genetic structure analysis of principal Pseudotaxus chienii natural populations in Zhejiang Province
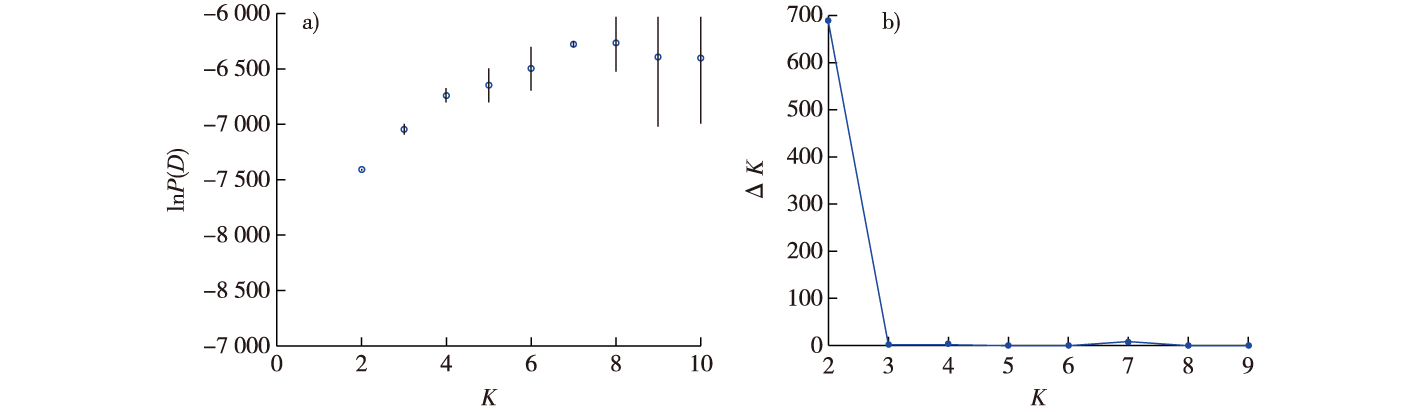
【Objective】The conservation of plant species with limited distribution and endangered status is a critical concern in biodiversity preservation. Among such species, Pseudotaxus chienii, a nationally protected plant species, has attracted significant attention due to its narrow habitat range and its vulnerability to extinction. As an endemic species primarily found in the mountainous regions of Zhejiang and Jiangxi provinces in China, P. chienii plays an important ecological role in maintaining forest biodiversity and stabilizing the local ecosystem. Unfortunately, over the past few decades, the natural populations of P. chienii have been severely affected by habitat destruction, fragmentation, and anthropogenic pressures, such as overharvesting and deforestation. These factors have contributed to a significant decline in both the population size and genetic diversity of this species. The research also sought to provide a deeper understanding of the evolutionary processes shaping the genetic makeup of P. chienii, especially in light of its restricted geographic range. By examining the genetic variation within and among populations, this study aimed to determine whether there are significant genetic differences that could have implications for long-term species survival. Additionally, the study aimed to investigate the extent of inbreeding and heterozygosity levels within these populations, which are important indicators of the overall fitness and adaptability of the species. The ultimate goal of the research was to generate critical genetic data that would serve as a foundation for future conservation and population restoration efforts, enabling the development of strategies that could enhance the species' genetic diversity and resilience in the wild.【Method】To achieve these objectives, a total of 133 individuals were collected from six natural populations of P. chienii located in different regions of Zhejiang Province (Fengyang Mountain Nature Reserve) and Quzhou City (Tianji Longmen Mountain Range). These populations were chosen for their geographic diversity, allowing for an examination of genetic variation across a range of environmental conditions and altitudes. In order to assess the genetic diversity and population structure of P. chienii, simple sequence repeat (SSR) molecular markers were employed. SSR markers are highly effective in detecting genetic variation and are widely used in plant population genetics studies. A total of 14 SSR primer pairs, known for their high polymorphism and ability to amplify variable loci, were selected for genotyping the 133 individuals. These markers were used to analyze a wide range of genetic parameters, including the number of alleles, the effective number of alleles (Ne), the Shannon's diversity index (I), and polymorphic information content (PIC). These parameters are commonly used to assess the level of genetic diversity within populations. Additionally, observed and expected heterozygosity (Ho and He) were calculated to evaluate the extent of genetic variation at the individual level, while fixation coefficients (F) and inbreeding coefficients (Fis) were determined to identify potential inbreeding and genetic drift within the populations. Furthermore, to explore the genetic structure of P. chienii, both principal coordinate analysis (PCoA) and unweighted pair group method with arithmetic mean (UPGMA) clustering were applied. These multivariate statistical methods allowed for the visualization of genetic relationships among individuals and provided insights into the clustering patterns of populations based on genetic similarities. By conducting these analyses, the study aimed to reveal any genetic subgroups within the species and identify the factors that contribute to the observed genetic structure. In summary, this study used advanced molecular techniques to investigate the genetic diversity and structure of P. chienii populations. By analyzing a large number of individuals from multiple natural populations, the study aimed to provide a comprehensive picture of the genetic variation within the species, and to understand the evolutionary and ecological factors that contribute to this variation.【Result】Significant differences were observed in tree height and diameter at breast height among the six populations of P. chienii, with the FYS2 population exhibiting the best growth status. Using 14 pairs of SSR primers with good polymorphism, genotyping of 133 P. chienii individuals identified a total of 102 alleles. The mean values of effective number of alleles (Ne), Shannon's diversity index (I), and polymorphic information content (PIC) were 4.69, 1.61 and 0.86, respectively. The average values of observed heterozygosity (Ho), expected heterozygosity (He), inbreeding coefficient (Fis) and fixation coefficient (F) were 0.34, 0.73, 0.54 and 0.58, respectively. Overall, the results indicated high genetic diversity in P. chienii populations, but there was a moderate level of heterozygote deficiency and obvious inbreeding within populations. Genetic structure analysis divided P. chienii individuals into two subgroups, which was also supported by PCoA and UPGMA clustering analysis.【Conclusion】The natural populations of P. chienii exhibit high levels of genetic diversity, with significant differences in genetic diversity among populations. Proximity-based clustering analysis reveals that geographically adjacent natural populations of P. chienii tend to cluster into the same subgroups, indicating that geographic variation is the primary factor influencing differences in the genetic structure in P. chienii.
Pseudotaxus chienii / nature populations / SSR markers / genetic diversity / genetic structure / Zhejiang Province
| [1] |
郑万钧, 傅立国. 中国植物志(第七卷)[M]. 北京: 科学出版社, 1978.
|
| [2] |
傅星, 南寅镐. 科尔沁沙地盐生草甸主要植物群落种群格局的研究[J]. 应用生态学报, 1992, 3(4):313-320.
|
| [3] |
徐晓婷, 杨永, 王利松. 白豆杉的地理分布及潜在分布区估计[J]. 植物生态学报, 2008, 32(5):1134-1145.
|
| [4] |
胡绍庆, 陈征海, 孙孟军. 浙江省白豆杉资源调查研究[J]. 浙江大学学报(农业与生命科学版), 2003, 29(1):97-102.
|
| [5] |
王康, 杨永. 红豆杉科白豆杉属植物的分类学研究[J]. 植物分类学报, 2007, 45(6):862-869.
|
| [6] |
杨旭, 于明坚, 丁炳扬, 等. 凤阳山白豆杉种群结构及群落特性的研究[J]. 应用生态学报, 2005, 16(7):1189-1194.
|
| [7] |
杨旭. 凤阳山自然保护区白豆杉种群生态学的研究[D]. 杭州: 浙江大学, 2005.
|
| [8] |
李红英, 李康琴, 胥猛, 等. 北美鹅掌楸EST-SSR跨属间通用性[J]. 东北林业大学学报, 2011, 39(2):28-30,42.
|
| [9] |
黄秦军, 苏晓华, 张香华. SSR分子标记与林木遗传育种[J]. 世界林业研究, 2002, 15(3):14-21.
|
| [10] |
苏应娟, 王艇, 郑博, 等. 根据cpDNA trnL-F非编码区序列变异分析黑桫椤海南和广东种群的遗传结构与系统地理[J]. 生态学报, 2004, 24(5):914-919.
|
| [11] |
章艺, 刘鹏. 衢州珍稀濒危植物区系的研究[J]. 井冈山师范学院学报, 2002, 23(5):30-33.
|
| [12] |
李小白, 向林, 罗洁, 等. 转录组测序(RNA-seq)策略及其数据在分子标记开发上的应用[J]. 中国细胞生物学学报, 2013, 35(5):720-726,740.
|
| [13] |
王希, 陈丽, 赵春雷. 利用MISA工具对不同类型序列进行SSR标记位点挖掘的探讨[J]. 中国农学通报, 2016, 32(10):150-156.
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
阮咏梅, 张金菊, 姚小洪, 等. 黄梅秤锤树孤立居群的遗传多样性及其小尺度空间遗传结构[J]. 生物多样性, 2012, 20(4):460-469.
|
| [22] |
杨梅, 张敏, 师守国, 等. 武当木兰种群遗传结构的ISSR分析[J]. 林业科学, 2014, 50(1):76-81.
|
| [23] |
罗芊芊, 李峰卿, 肖德卿, 等. 两个南方红豆杉天然居群的交配系统分析[J]. 南京林业大学学报(自然科学版), 2023, 47(5):80-86.
|
| [24] |
程建慧, 廖荣俊, 宋其岩, 等. 基于SSR标记分析浙西南香椿天然居群的遗传多样性[J]. 中南林业科技大学学报, 2024, 44(2):146-156,165.
|
| [25] |
臧明月, 李璇, 方炎明. 基于SSR标记的白栎天然居群遗传多样性分析[J]. 南京林业大学学报(自然科学版), 2021, 45(1):63-69.
|
| [26] |
胡绍庆. 浙江省白豆杉植物群落区系研究[J]. 浙江林业科技, 2001, 21(5):59-62.
|
| [27] |
林雪莹. 水松EST-SSR分子标记开发及群体遗传变异分析[D]. 长沙: 中南林业科技大学, 2018.
|
| [28] |
|
| [29] |
陈文文. 水杉(Metasequoia glyptostroboides)自然种群交配系统和扩散格局研究[D]. 上海: 华东师范大学, 2016.
|
| [30] |
邓莉丽, 刘青华, 周志春, 等. 抗松材线虫病马尾松种质资源遗传多样性分析及核心种质构建[J]. 浙江农林大学学报, 2024, 41(1):67-78.
|
| [31] |
魏利斌, 苗红梅, 李春, 等. 芝麻SNP和InDel标记遗传多样性、群体结构及连锁不平衡分析[J]. 分子植物育种, 2017, 15(8):3070-3079.
|
| [32] |
刘珊廷, 易显荣, 周民武, 等. 广西梨种质资源遗传多样性和群体结构分析[J]. 果树学报, 2024, 41(3):379-391.
|
| [33] |
杜康, 王加焕, 李俊恒, 等. 毛白杨耐寒种质资源遗传鉴定及评价[J]. 北京林业大学学报, 2023, 45(12):59-67.
|
| [34] |
罗芊芊. 南方红豆杉天然居群及家系遗传变异研究[D]. 长沙: 中南林业科技大学, 2020.
|
| [35] |
张俊红, 王洋, 周生财, 等. 闽楠群体遗传结构分析与核心种质库构建[J]. 林业科学, 2024, 60(1):68-79.
|
| [36] |
|
| [37] |
中国科学院华南研究所华南植物园, 广东省野生动植物保护管理办公室. 广东珍稀濒危植物[M]. 北京: 科学出版社, 2003.
South China Botanical Garden, South China Institute, Chinese Academy of Sciences,Guangdong Provincial Wildlife Conservation and Management Office. Rare and endangered plants in Guangdong[M]. Beijing: Science Press, 2003.
|
衢州市柯城区林业技术推广中心刘汝明和中国林业科学研究院亚热带林业研究所金国庆副研究员在材料收集与研究工作中给予了帮助。
/
| 〈 |
|
〉 |





